

Hair Restore Ultra Scalp Solution
Product Overview
Minoxidil
Oral minoxidil (Loniten) is an antihypertensive agent; topical minoxidil (Rogaine) is used for alopecia. Topical minoxidil is likely effective in producing moderate hair growth in approximately 30% of men and 60% of women with common hereditary hair loss.[1] Lesser growth or a halt in the worsening of alopecia are also frequent outcomes.[2] Due to its potency and adverse reactions, oral minoxidil is used mainly for patients with severe, drug-resistant forms of hypertension. Tolerance to prolonged therapy with oral minoxidil does not appear to be a problem. Although the oral dosage form was originally approved in October 1979 for use in hypertension, minoxidil was first discovered in 1965. In August 1988 the topical formulation was approved for of alopecia. After declining Upjohn permission to market topical minoxidil as a non-prescription drug in July 1994, the 2% topical solution was subsequently approved for over-the-counter use in men with alopecia in February 1996. In September 1996, Pharmacia and Upjohn petitioned the FDA to increase the topical solution formulation from 2% to 5%; the higher-strength solution has been shown to elicit a more rapid hair growth response (8 weeks vs. 16 weeks) and to regrow an average of 45% more hair than Rogaine Regular Strength.[2] The 5% topical solution (Rogaine Extra Strength) for common hereditary hair loss was approved FDA in November 1997. Approval to market 2% topical solution to women was granted October 1996. Minoxidil foam (Men’s Rogaine Foam) was approved for men in January 2006; potential advantages over the solution formulation include the absence of propylene glycol (potential irritant), ability to limit spread of medication, and less time to dry after application.[3]
Azelaic Acid
Azelaic acid is a topical anti-acne agent. Azelaic acid is a naturally occurring dietary constituent (whole grain and animal products) and can be formed endogenously from longer-chain dicarboxylic acids, metabolism of oleic acid, and omega-oxidation of monocarboxylic acids. Azelaic acid is used as a treatment for acne vulgaris and is one of the leading treatments for this condition in Europe where it has been marketed as a 20% cream since 1989 by Schering. The drug is also effective for reducing the number of inflammatory pustules and papules associated with rosacea. Reduction in erythema may occur; however, efficacy has not been established for erythema associated with rosacea without papules and pustules. Azelaic acid has been administered orally and intravenously; however, only a topical formulation is marketed in the United States. The drug was originally FDA approved as a 20% cream for the treatment of acne vulgaris in September 1995; several brand name creams are now available. A 15% topical gel and foam were FDA-approved for the treatment of rosacea in December 2002 and July 2015, respectively.[4][5][6]
Finasteride
Finasteride is a 5-alpha reductase inhibitor used to treat symptomatic benign prostatic hyperplasia (BPH), a condition found in the majority of men over the age of 50. Finasteride has been shown to increase and maintain maximum urine flow rate in men with BPH, although less than 50% of men show improvement despite a reduction in prostate size. In a typical patient undergoing treatment for BPH with finasteride (>= 6 months), a 50% decrease in serum PSA concentrations can be expected; however, individual patients may experience varying decreases in PSA values. During treatment, serum PSA concentrations may decrease even in the presence of prostate cancer. If clinicians use serum PSA concentrations as an aid in the detection of prostate cancer in men receiving finasteride, values should be doubled for comparison with normal ranges in untreated men. Any increase from baseline, even if the value is within the normal range for untreated men, may signal the presence of prostate cancer. If clinicians elect to use percent free PSA (free to total PSA ratio) as a marker, no adjustment in PSA values appear to be necessary as the value is not significantly decreased by finasteride.[7] In June 2011, a review of two large, randomized controlled trials, the Prostate Cancer Prevention Trial (PCPT) and the Reduction by Dutasteride of Prostate Cancer Events (REDUCE) trial prompted the FDA to alert healthcare professionals of the potential risk of an increased incidence of high-grade prostate cancer in patients receiving finasteride or dutasteride treatment. Results from the PCPT trial showed that men receiving finasteride had a 26% decreased risk of being diagnosed with prostate cancer overall when compared to placebo (p < 0.0001); however, the risk reduction was limited to Gleason score (GS) <= 6 cancers. There was an increased incidence of GS 8-10 prostate cancers with finasteride compared to placebo (1.8% vs. 1.1%, respectively).[8][9] Finasteride is also used for treating hair loss in men and has been shown to be effective for mild to moderate hair loss of the vertex and anterior mid-scalp area; efficacy in bitemporal recession has not been established. Finasteride (Proscar) was approved by the FDA in June 1992 for the treatment of BPH. Another finasteride oral dosage form, Propecia, was approved by the FDA in December 1997 for the treatment of male pattern baldness (i.e., androgenetic alopecia). Finasteride is also used investigationally as an alternative agent for treating hirsutism.
Ketoconazole
Ketoconazole is an imidazole antifungal agent and is available as an oral tablet and various topical formulations. Oral ketoconazole has been associated with fatal hepatotoxicity, adrenal gland suppression, and harmful drug interactions. Due to the potential for severe adverse events, oral ketoconazole should only be used to treat serious blastomycosis, histomycosis, coccidioidomycosis, paracoccidioidomycosis, and chromomycosis infections when no other antifungal therapies are available. The topical formulations are indicated for the treatment of tinea corporis, tinea cruris, tinea pedis, tinea versicolor, mucocutaneous candidiasis, seborrheic dermatitis, and dandruff. Ketoconazole was FDA-approved in 1981.[10][11][12][13][14]
Minoxidil
Minoxidil has a direct vasodilatory effect on arterial smooth muscle, causing a reduction in peripheral resistance and blood pressure. Minoxidil does not exhibit CNS or adrenergic neuronal blocking effects; minoxidil retains its activity despite adrenergic denervation. Cyclic adenosine monophosphate (cAMP) may contribute to relaxation of vascular smooth muscle. Minoxidil-induced delay in the hydrolysis of cAMP via inhibition of phosphodiesterase may contribute to the drug’s vasodilatory action.
All direct vasodilators produce a sympathetic response including an increase in heart rate, stroke volume, and cardiac output, and a marked increase in plasma renin activity, which, in turn, leads to increased sodium and water retention. This increased renin release is believed to be partially mediated by the beta-adrenergic system. These compensatory responses tend to diminish the hypotensive effects of minoxidil. Additional therapeutic effects can be achieved by using a beta-blocker to offset the predictable sympathetic stimulation caused by minoxidil. Methyldopa may be used if beta-blocker therapy is contraindicated; however, because of its delay in onset, methyldopa must be initiated 24 hours prior to initiating minoxidil. Vasodilator-induced fluid retention is somewhat related to the potency of the vasodilator. Due to its potency, fluid retention occurs routinely with minoxidil. Often, this fluid retention requires concomitant use of loop diuretics (see Adverse Reactions). Triple-drug therapy consisting of a loop diuretic, beta-blocker, and minoxidil produces prompt, sustained reduction in blood pressure in patients with severe hypertension.[15][16]
Minoxidil preferentially dilates arterioles; therefore, postural hypotension may occur during therapy. As an antihypertensive, minoxidil does not lead to improvements in LVH. Minoxidil may actually worsen LVH, potentially due to reflex tachycardia and sympathetic stimulation, which may counteract the benefits of afterload reduction.[17] Minoxidil does not affect glucose tolerance or serum lipids.
The exact mechanism responsible for minoxidil-induced hair growth is not known, but appears to be independent of vasodilation.[18] While systemic therapy will stimulate hair growth, topical therapy usually does not cause hypotension. Current evidence suggests the primary action of topical minoxidil is to decrease the latent period of the hair cycle. The latent period (the time between shedding of telogen hair and the onset of the next anagen) is typically prolonged in male pattern balding; however, this effect has not been demonstrated in balding females.[19] Calcium may also be involved in the process of hair regrowth. In the presence of calcium, epidermal growth factor (EGF) inhibits hair growth. The entry of calcium into a hair cell is opposed by potassium channel openers, such as minoxidil; therefore, EGF-induced inhibition of hair will be opposed by the action of minoxidil, and hair will grow more proficiently. Biopsy specimens have not demonstrated evidence of new follicle formation with the use of minoxidil.[1] Furthermore, minoxidil appears to affect only suboptimal follicles with no further stimulation of normal hair follicles.[18] Minoxidil also may alter the metabolism of androgens in the scalp. Minoxidil increases 17 beta-hydroxylated dehydrogenase activity by almost 40% in dermal papilla cells of a balding scalp, whereas the effect is much less in a nonbalding scalp. Whether this modification in testosterone metabolism of cells of a balding scalp is related to the therapeutic effect of minoxidil is unknown.[20]
Azelaic Acid
The efficacy of azelaic acid in acne vulgaris is due to an antimicrobial effect and an antikeratinizing effect on the follicular epidermis. The antimicrobial effects of azelaic acid involves inhibition of synthesis of microbial cellular proteins; the exact mechanism of action is unknown. Azelaic acid possesses bacteriostatic properties against a variety of aerobic microorganisms, especially Staphylococcus epidermidisand Propionibacterium acnes which are known to be elevated in acne-bearing skin; at high concentrations, azelaic acid is bactericidal against S. epidermidis and P. acnes. By reducing the concentration of bacteria present on the skin, azelaic acid decreases the inflammation associated with acne lesions. Azelaic acid may also possess a direct antiinflammatory effect by scavenging oxygen radicals. The antikeratinizing effects of azelaic acid may be due to decreased synthesis of filaggrin (keratin filament aggregating protein). By inhibiting filaggrin, azelaic acid may normalize the keratinization of the follicle and produce a reduction in noninflamed acne lesions. Azelaic acid does not affect sebum excretion.
The mechanism of action that results in the efficacy of azelaic acid in acne rosacea is not clear; clinical studies suggest interference with the pathogenic effects in rosacea. Anti-inflammatory effects have been noted in vitro.
The antiproliferative and cytotoxic actions of azelaic acid may be due to reversible inhibition of a variety of oxidoreductive enzymes including DNA polymerase, tyrosinase, and mitochondrial enzymes of the respiratory chain. At the cellular level, azelaic acid causes mitochondrial swelling and accumulation of cytoplasmic lipid droplets. Azelaic acid has shown efficacy in treating such conditions as lentigo maligna, cutaneous malignant melanoma, and melasma (chloasma). When azelaic acid is applied topically in these conditions, there is a reduction in epidermal melanogenesis and replacement of abnormal melanocytes by normal cells; flattening of nodular areas may also occur. Hyperactive and malignant melanocytes are much more susceptible to the effects of azelaic acid than are normal melanocytes.
Finasteride
Finasteride is a synthetic 4-aza analog of testosterone that acts as a competitive, specific inhibitor of type II 5-alpha-reductase, an intracellular enzyme that converts testosterone to the potent androgen 5-alpha-dihydrotestosterone (DHT). The type II 5alpha-reductase isozyme is primarily found in prostate, seminal vesicles, epididymides, and hair follicles, as well as liver. The type II isozyme is responsible for two-thirds of circulating DHT. DHT is the primary androgen that stimulates the development of prostate tissue. When used for the treatment of benign prostatic hyperplasia, as the enzymatic conversion from testosterone to DHT is inhibited, a desirable reduction in prostate hypertrophy is achieved, and urine flow should be improved. In male pattern hair loss, the balding scalp contains miniaturized hair follicles and increased amounts of DHT compared with hairy scalp. Finasteride decreases scalp and serum DHT concentrations, thus interrupting a key factor in the development of androgenetic alopecia in those patients genetically predisposed. Finasteride does not appear to affect circulating concentrations of cortisol, estradiol, prolactin, thyroid-stimulating hormone, thyroxine or cholesterol. Research to date also suggests that finasteride does not affect the hypothalamic-pituitary-testicular-axis.
Ketoconazole
Like other azole antifungals, ketoconazole exerts its effect by altering the fungal cell membrane. Ketoconazole inhibits ergosterol synthesis by interacting with 14-alpha demethylase, a cytochrome P-450 enzyme that is necessary for the conversion of lanosterol to ergosterol, an essential component of the membrane. In contrast, amphotericin B binds to ergosterol after it is synthesized. Inhibition of ergosterol synthesis results in increased cellular permeability, which causes leakage of cellular contents. Ketoconazole does not appear to have the same effects on human cholesterol synthesis. Other antifungal effects of azole compounds have been proposed and include: inhibition of endogenous respiration, interaction with membrane phospholipids, and inhibition of yeast transformation to mycelial forms.[21] Other mechanisms may involve inhibition of purine uptake and impairment of triglyceride and/or phospholipid biosynthesis.[22] At higher concentrations, ketoconazole may have a direct physiochemical effect on the fungal cell membrane, which leads to a fungicidal action.
Ketoconazole possesses actions that may make it useful in conditions other than fungal infections. Ketoconazole can inhibit sterol synthesis in humans including the synthesis of aldosterone, cortisol, and testosterone. Ketoconazole’s effects on testosterone synthesis occur at lower doses than do the effects on cortisol synthesis; doses of of 200-400 mg/day can inhibit testosterone secretion and doses of 400-600 mg/day have been shown to inhibit cortisol synthesis.[23][24] Ketoconazole acts at many of same steps as metyrapone and, in some sites, has been shown to be a more potent inhibitor.[23] Both ketoconazole and metyrapone affect multiple steps in the steroid-synthesis pathway, while finasteride appears to work at a single site. Ketoconazole has been used successfully for treating advanced prostate cancer.[25] Finally, ketoconazole is a known potent inhibitor of thromboxane synthesis and has been used clinically to prevent ARDS in patients at high risk of this syndrome.[26]
Minoxidil
Minoxidil has been reported to produce cardiac lesions in animals. Some lesions are characteristic of other drugs that can cause tachycardia and/or hypotension (e.g., isoproterenol, hydralazine). These effects are more likely to occur in patients with compromised renal function and in patients with connective tissue disease, uremic syndrome, CHF, or minoxidil-induced fluid retention.
Systemic minoxidil is a potent vasodilator with potential to produce hypotension and reflex tachycardia; serious complications may occur. Minoxidil is relatively contraindicated in patients with cardiac disease (including angina, coronary artery disease, recent or acute myocardial infarction), or cerebrovascular disease because a reflex increase in heart rate and decrease in blood pressure can exacerbate these conditions. Minoxidil is relatively contraindicated in patients with coronary insufficiency, including angina, to avoid the risk of reflex tachycardia and angina exacerbation. Minoxidil may cause pericardial effusion which occasionally may progress to cardiac tamponade. Reserve oral minoxidil for hypertension in patients who do not respond adequately to maximum therapeutic doses of a diuretic (loop diuretic suggested) concurrently with 2 other antihypertensive agents. In experimental animals, minoxidil has been shown to induce several types of myocardial lesions as well as other adverse cardiac effects. Minoxidil must be administered under close supervision, usually in combination with therapeutic doses of a beta-blocker to prevent reflex tachycardia and increased myocardial workload. Minoxidil is often given with a diuretic (preferably a diuretic which acts within the ascending limb of the loop of Henle) to prevent fluid accumulation and peripheral edema. When first administering minoxidil to patients with malignant hypertension and those already receiving guanethidine to avoid rapid or large orthostatic reductions in blood pressure, minoxidil use requires a specialized care setting, specifically hospitalization. Although minoxidil does not directly cause orthostatic hypotension, administration to patients receiving guanethidine can result in profound orthostatic effects. When possible, guanethidine should be discontinued well before minoxidil is initiated. Otherwise, minoxidil therapy should be started in the hospital; the patient should remain hospitalized until the risk of excessive orthostatic effects is minimized and the patient is able to avoid activities that induce orthostatic hypotension.
Minoxidil is relatively contraindicated in patients with renal disease, preexisting pulmonary hypertension, or chronic congestive heart failure not secondary to hypertension because the drug can cause an increase in pulmonary artery pressure, which could be detrimental to these patients. Use of minoxidil has been associated with the development of pericardial effusion and tamponade in some patients, and it may be more likely to occur in patients with renal disease. Since approximately only 10% of active drug is eliminated unchanged via the kidneys, minoxidil can be used safely in patients with renal impairment. Renal elimination, however, may be reduced and dosage adjustment may be necessary. Avoid use of minoxidil in patients with severe renal failure (CrCl < 10 ml/min). Minoxidil is contraindicated in patients with pheochromocytoma because the hypotensive effects of the drug can stimulate catecholamine secretion. Systemic effects resulting from topically administered minoxidil are unlikely but theoretically could occur if the drug is overused. Skin abrasion or irritations, such as excoriations, psoriasis, or sunburn, can increase the systemic absorption of topically administered minoxidil. Reported clinical experience has not identified differences in responses in geriatric adults vs. younger adult patients. In general, systemic dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.[27] Topical minoxidil use carries no special precaution in the elderly, but any patient experiencing dizziness or faintness should discontinue topical use.[1] The federal Omnibus Budget Reconciliation Act (OBRA) regulates medication use in residents of long-term care facilities (LTCFs). According to OBRA, antihypertensive regimens should be individualized to achieve the desired outcome while minimizing adverse effects. Antihypertensives may cause dizziness, postural hypotension, fatigue, and there is an increased risk for falls. There are many drug interactions that can potentiate the effects of antihypertensives. Some agents require a gradual taper to avoid adverse consequences caused by abrupt discontinuation.[28] Azelaic Acid Azelaic acid products that contain propylene glycol should be avoided in patients with a known propylene glycol hypersensitivity; avoid use in patients hypersensitive to any other ingredients of the particular formulation prescribed.[4] Azelaic acid has not been well-studied in patients with dark complexions and should be used cautiously in these patients to avoid hypopigmentation.[4][6] An occlusive dressing should not be used with azelaic acid. Avoid ocular exposureand accidental exposure/contact with the mouth and other mucous membranes. If contact with the eye(s) occur, the eye(s) should be washed with large amounts of water; patients should contact their physician if ocular irritation persists. The safety and effectiveness of azelaic acid cream and gel formulations in neonates, infants, and children under 12 years of age have not been established. The foam formulation is not approved for use in pediatric patients less than 18 years of age.[6] Do not apply azelaic acid to areas affected by herpes labialis; exacerbations of herpes infection have been reported. Worsening or deterioration of asthma has been observed in patients treated with azelaic acid. Instruct drug recipients to contact their physician if signs of an asthma exacerbation (i.e., dyspnea, wheezing) develop during therapy.[4] Finasteride Finasteride is not indicated for use in adolescents, children, or infants. Safety and effectiveness have not been established in pediatric patients under 18 years of age.[7][29] Finasteride should be used with caution in patients with hepatic disease, since finasteride is metabolized extensively in the liver. Data are lacking regarding the incidence of adverse effects or drug accumulation in patients with hepatic impairment. Finasteride reduces total serum prostate specific antigen (PSA). In a typical patient undergoing treatment for BPH with finasteride (>= 6 months), a 50% decrease in serum PSA concentrations can be expected; however, individual patients may experience varying decreases in PSA values. During treatment, serum PSA concentrations may decrease even in the presence of prostate cancer. If clinicians use serum PSA concentrations as an aid in the detection of prostate cancer in men receiving finasteride, values should be doubled for comparison with normal ranges in untreated men. Any increase from baseline, even if the value is within the normal range for untreated men, may signal the presence of prostate cancer. If clinicians elect to use percent free PSA (free to total PSA ratio) as a marker, no adjustment in PSA values appear to be necessary as the value is not significantly decreased by finasteride.[7] In June 2011, a review of two large, randomized controlled trials, the Prostate Cancer Prevention Trial (PCPT) and the Reduction by Dutasteride of Prostate Cancer Events (REDUCE) trial prompted the FDA to alert healthcare professionals of the potential risk of an increased incidence of high-grade prostate cancer in patients receiving finasteride or dutasteride treatment. Results from the PCPT trial showed that men receiving finasteride had a 26% decreased risk of being diagnosed with prostate cancer when compared to placebo (p < 0.0001); however, the risk reduction was limited to Gleason score (GS) <= 6 cancers. There was an increased incidence of GS 8-10 prostate cancers with finasteride compared to placebo (1.8% vs. 1.1%, respectively).[8][9] Therefore, in initiating or continuing treatment with finasteride, clinicians should weigh the known benefits of treatment against the potential risk and be aware that finasteride may increase the risk of high-grade prostate cancer. Further, lower urinary tract symptoms of BPH can be indicative of other urological diseases, including prostate cancer. Patients should be assessed to rule out other urological diseases prior to treatment with finasteride. Patients with a large residual urinary volume and/or severely diminished urinary flow may not be good candidates for 5-alpha-reductase inhibitor therapy and should be carefully monitored for urinary tract obstruction.[7]
Men treated with finasteride should refrain from blood donation while taking finasteride. The purpose of this is to prevent administration of finasteride to a pregnant female transfusion recipient.
Clinical efficacy studies of finasteride for hair loss did not include subjects aged 65 and over. Based on the pharmacokinetics of finasteride 5 mg, no dosage adjustment is necessary in the geriatric patient. However, the efficacy of finasteride for hair loss in the elderly has not been established.
The clinical significance of finasteride’s effect on semen characteristics for an individual male patient’s fertility is not known; consider the potential effects on semen when assessing a male with infertility. Finasteride may cause spermatogenesis inhibition or oligospermia, decreased sperm motility, or decreased semen volume. In a 52-week, randomized, double-blind, placebo-controlled study in healthy men, finasteride (5 mg PO once daily) significantly decreased total sperm count (-34.3%) compared to baseline at 26 weeks but not at 52 weeks or at the 24-week follow-up. Semen volume was decreased at 52 weeks for finasteride (-14.5%), but the effect was not statistically significant. Sperm concentration was decreased by finasteride (-7.4%) but was not significant for either drug. Significant reductions of 6 to 12% in sperm motility were observed during treatment. Sperm morphology was not affected. One subject taking finasteride had decreases in sperm count of more than 90% of baseline values at 52 weeks; partial recovery was noted at the 24-week follow-up. During post marketing surveillance, male infertility and/or poor seminal quality following treatment discontinuation have been reported. It should be noted that normalization or improvement of seminal quality has also been reported after discontinuation of finasteride.[7][29][30]
Ketoconazole
Due to its potent inhibition of the hepatic isoenzyme CYP3A4, oral ketoconazole coadministration with other drugs metabolized by CYP3A4 should be done with extreme caution, if at all. Ketoconazole can cause elevated plasma concentrations of selected drugs metabolized via CYP3A4 which may prolong the QT interval, sometimes resulting in life-threatening ventricular arrhythmias such as torsade de pointes; use of ketoconazole with such drugs is contraindicated. Oral ketoconazole may also inhibit the metabolism of many other drugs, which could result in serious and potentially life-threatening adverse reactions, and use with selected drugs is also contraindicated.[10] Due to the potential for harmful drug interactions and other serious adverse effects, oral ketoconazole should only be used to treat serious fungal infections when no other antifungal therapies are available.[13][14] Systemic ketoconazole can prolong the QT interval. Use ketoconazole tablets with caution in patients with cardiac disease or other conditions that may increase the risk of QT prolongation including cardiac arrhythmias, congenital long QT syndrome, heart failure, bradycardia, myocardial infarction, hypertension, coronary artery disease, hypomagnesemia, hypokalemia, hypocalcemia, or in patients receiving medications known to prolong the QT interval or cause electrolyte imbalances. Females, older adult patients, patients with diabetes mellitus, thyroid disease, malnutrition, alcoholism, or hepatic disease may also be at increased risk for QT prolongation.[10][31][32][33][34][35][36]
Ketoconazole oral tablets may cause adrenal insufficiency at doses of 400 mg/day and higher in adults. This effect is not shared with other azole antifungals. The recommended dose of 200 to 400 mg daily in adults should not be exceeded. Adrenal function should be monitored in patients with adrenal insufficiency or with borderline adrenal function and in patients under prolonged periods of stress (major surgery, intensive care, etc.).[10] Due to the risk of adrenal insufficiency and other serious adverse effects, oral ketoconazole should only be used to treat serious fungal infections when no other antifungal therapies are available.[13][14]
Ketoconazole should be used with caution in patients with known azole antifungals hypersensitivity. Hypersensitivity reactions may be due to the various vehicles present in the different ketoconazole formulations. Ketoconazole may have a cross sensitivity with other azole derivatives such as itraconazole, fluconazole, clotrimazole, and miconazole. In rare cases, patients receiving ketoconazole have reported hypersensitivity reactions and even anaphylaxis. Ketoconazole is contraindicated in patients who have previously demonstrated these reactions.
Avoid accidental ocular exposure of topical ketoconazole products. If ocular exposure occurs, treat by immediate flushing the affected eye with cool, clean water. Contact an ophthalmologist if eye irritation persists.
Some topical ketoconazole products are flammable. Due to the alcohol content of ketoconazole topical gel (e.g., Xolegel gel) and the alcohol, butane, and propane content of ketoconazole topical foam (e.g., Extina foam), avoid fire, flame, or tobacco smoking during and immediately after the application of these ketoconazole products.
Dizziness or drowsiness occurs in some patients receiving systemic ketoconazole. Patients should be careful driving or operating machinery if they have these reactions.
Due to the risk of severe drug interactions and other serious adverse effects with ketoconazole oral tablets, ketoconazole oral tablets should not be a first-line treatment for any fungal infection in the geriatric patient.[10][13] Systemic ketoconazole can prolong the QT interval. Geriatric patients may be at increased risk for QT prolongation and for serious drug-drug interactions that may increase the risk QT prolongation risk or may increase the risk for other serious side effects.[10][31][32][33][34][35][36] The federal Omnibus Budget Reconciliation Act (OBRA) regulates medication use in residents of long-term care facilities (LTCFs). According to OBRA, systemic azole antifungals should be used in the lowest possible dose for the shortest possible duration, particularly in patients receiving other medications known to interact with these medications. Increased monitoring may be required to identify and minimize the toxicity of warfarin, phenytoin, theophylline, or sulfonylureas when an azole antifungal is co-administered; other medications such as rifampin and cimetidine may decrease the therapeutic effect of the antifungal. Some drug-drug combinations may be contraindicated. OBRA guidelines caution that azole antifungals may cause hepatotoxicity, headaches, and GI distress.[28]
The safety and efficacy of oral ketoconazole have not been established in neonates, infants, or children under 2 years of age.[10] Topical products (e.g., shampoo, cream) have been used in pediatric patients off-label but are not FDA-approved for use in pediatric patients; the safety and efficacy of ketoconazole topical foam and gel products have not been established in pediatric patients less than 12 years old.[11][37][38][39]
Minoxidil
The adverse reaction profile for minoxidil depends upon its use. Systemic adverse reactions are unlikely from topical administration. Placebo-controlled trials with topical minoxidil only showed an increase in dermatological effects from the active drug.
Minoxidil is a peripheral vasodilator. All direct vasodilators produce a marked increase in plasma renin activity, which leads to water and sodium retention and sometimes congestive heart failure. This renin release is believed to be partially mediated by the beta-adrenergic system. The degree of fluid retention is somewhat related to the potency of the vasodilator. Due to its potency, fluid retention (edema) occurs routinely with oral minoxidil and usually requires concomitant administration of a loop diuretic. Without a diuretic, rapid fluid retention can occur within a few days of minoxidil therapy. Temporary edema occurred in 7% of patients who were not edematous when minoxidil was initiated.[27] Ascites also has been reported. A restricted dietary intake of sodium can minimize fluid retention and resultant peripheral edema. Rarely, fluid retention is refractory to diuresis and discontinuation of minoxidil is required. Vasodilation may also produce headache.
Minoxidil causes reflex tachycardia; sinus tachycardia may occur. Angina may become apparent, or worsen, secondary to increased myocardial oxygen demand associated with tachycardia and increased cardiac output. Tachycardia and subsequent angina usually can be prevented with the coadministration of a beta-blocker or other sympathetic nervous system suppressant.[27]
Minoxidil has been shown to transiently lower hematocrit, hemoglobin, and erythrocyte count by approximately 7%. Serum creatinine and BUN also have been shown to increase an average of 6% in patients on minoxidil therapy. Increases in alkaline phosphatase, without other evidence of hepatic abnormality, also has been reported. During the course of therapy, these laboratory abnormalities have been shown to return to pretreatment values. Thrombocytopenia and leukopenia also have been reported.[27]
Hypertrichosis (elongation, thickening, and enhancement of fine body hair), without evidence of virilism or endocrine abnormalities, is an embarrassing adverse effect that often occurs with oral minoxidil. This effect is usually evident within 3-6 weeks of therapy and occurs on the temples, between the eyebrows, or in the sideburn area. Hair growth also can appear on the arms, legs, and scalp. It is reversible following discontinuation of the drug.[27]
Oral minoxidil has occasionally been associated with appearance of a bullous rash and Stevens-Johnson syndrome. Topical minoxidil therapy produces local dermatological reactions including contact dermatitis, local burning, pruritus, erythema, or xerosis. Many other adverse effects have been reported during administration of topical minoxidil preparations, but none has been directly attributed to the drug.[27]
Gastrointestinal adverse effects associated with orally administered minoxidil include nausea and vomiting.[27]
Azelaic Acid
Most side effects occurring with the use of azelaic acid are dermatologic in nature and mild in severity. These effects include burning sensation or stinging (1-6.2%), paresthesias or tingling (1-6.2%), pruritus (1-5%), xerosis (dry skin, < 5%), erythema (< 2%), skin irritation (< 2%), contact dermatitis (< 1%), rash (unspecified) (< 1%), peeling (< 1%), dermatitis (< 1%), and edema (< 1%). In patients with dark complexions, skin hypopigmentation may occur. The following additional adverse reactions have been reported rarely: vitiligo depigmentation, small depigmented spots, hypertrichosis, reddening (signs of keratosis pilaris), and exacerbation of recurrent herpes viral infection (i.e., herpes labialis).[4][5][6]
Post-marketing use of azelaic acid has been associated with the development of hypersensitivity reactions (including angioedema, ocular inflammation, facial swelling, and urticaria) and asthma exacerbation (i.e., dyspnea, wheezing). In addition, cases of iridocyclitis, or inflammation of the iris, have been noted following accidental exposure of the eye to the topical gel. Due to the voluntary nature of post-marketing reports, neither a frequency nor a definitive causal relationship can be established.[4]
Finasteride
Adverse reactions to finasteride are generally mild and transient. In a long-term (4 years) clinical trial in men with benign prostatic hypertrophy (BPH), the most frequently reported adverse reactions to finasteride were related to sexual function. At 1 year, the adverse reactions reported to be drug-related were impotence (erectile dysfunction), decreased libido, decreased ejaculate volume, ejaculation dysfunction, breast enlargement, breast tenderness (mastalgia), and rash (unspecified). There was no significant difference between finasteride and placebo in the incidences of impotence, decreased libido, and ejaculation dysfunction in years 2 to 4 of the study. However, during post marketing surveillance, continued erectile dysfunction, orgasm dysfunction or other orgasm disorders, and ejaculation dysfunction following treatment discontinuation have been reported.[7][29] From June 1992, when finasteride was approved, until February 1995, the FDA received reports of gynecomastia in 214 men (median age: 71 yrs). Most were taking a dose of 5 mg/day PO. Gynecomastia has been the most frequently reported adverse effect of this drug since it was marketed. The onset of gynecomastia ranged from 14 days to 2.5 years (median: 180 days). Thirty percent had unilateral gynecomastia, 25% had bilateral involvement, and, in the remainder of reports, this information was not specified. Twenty-seven percent of patients were also taking other medications that are known to cause gynecomastia. Gynecomastia resolved either completely or partially in 80% of subjects after finasteride was discontinued, however, in at least 2 cases, a new primary malignancy of primary intraductal breast cancer subsequently developed.[48] In a 4 to 6 year trial where patients were randomized to receive finasteride 5 mg/day, doxazosin 4 or 8 mg/day, a combination of the two drugs, or placebo, four patients reported breast cancer as an adverse experience; three of the patients were receiving finasteride therapy and one patient was receiving combination therapy. In addition, male breast cancer has been reported during post-marketing experience. Other post-marketing adverse reactions have included depression, testicular pain that continued after discontinuation of treatment, and hypersensitivity reactions including pruritus, urticaria, and angioedema (including swelling of the lips, tongue, throat, and face).[7]
In controlled trials of finasteride for the treatment of male pattern hair loss, 1.4% of patients discontinued therapy due to adverse events, compared with 1.6% of placebo-treated patients. Discontinuation of therapy because of a drug-related sexual adverse experience occurred in 1.2% of patients on finasteride and 0.9% of patients on placebo. The following adverse events were reported as at least possibly drug-related in finasteride-treated patients: libido decrease (1.8%), impotence (1.3%), and ejaculation disorder (1.2%), primarily decreased ejaculate volume. The incidence of each of the above adverse effects decreased to <= 0.3% by the fifth year of treatment. During post marketing surveillance, decreased libido and libido disorders that continued after discontinuation of treatment was reported.[7]
Finasteride may cause spermatogenesis inhibition or oligospermia, decreased sperm motility, or decreased semen volume. The clinical significance of finasteride’s effect on semen characteristics for an individual male patient’s fertility is not known; consider the potential effects on semen when assessing a male with infertility. In a 52-week, randomized, double-blind, placebo-controlled study in healthy men, finasteride (5 mg PO once daily) significantly decreased total sperm count (-34.3%) compared to baseline at 26 weeks but not at 52 weeks or at the 24-week follow-up. Semen volume was decreased at 52 weeks for finasteride (-14.5%), but the effect was not statistically significant. Sperm concentration was decreased by finasteride (-7.4%) but was not significant for either drug. Significant reductions of 6 to 12% in sperm motility were observed during treatment. Sperm morphology was not affected. One subject taking finasteride had decreases in sperm count of more than 90% of baseline values at 52 weeks; partial recovery was noted at the 24-week follow-up. During post marketing surveillance, male infertility and/or poor seminal quality following treatment discontinuation have been reported. It should be noted that normalization or improvement of seminal quality has also been reported after discontinuation of finasteride.[7][29][30]
By inhibiting the conversion of testosterone to DHT, finasteride and other 5-alpha-reductase inhibitors have the ability to cause teratogenesis, specifically abnormalities in the external genitalia of the male fetus (e.g., hypospadias).[7]
Ketoconazole
Approximately 3% of patients receiving oral ketoconazole experience episodes of nausea and vomiting. This usually is transient and may improve when ketoconazole is given with food, which also can increase oral bioavailability. Other ketoconazole-associated gastrointestinal adverse reactions include abdominal pain and diarrhea which were reported in 1.2% and less than 1% of patients, respectively.[10] Cases of cheilitis have been noted during postmarketing use of ketoconazole topical foam. Due to the voluntary nature of postmarketing reports, neither a frequency nor definitive causal relationship can be established.[38]
Ketoconazole can inhibit testosterone secretion at doses of 200-400 mg/day and can inhibit cortisol synthesis in doses of 400-600 mg/day.[23][24] Inhibition of testosterone synthesis has led to cases of gynecomastia and impotence (erectile dysfunction) in < 1% of men. Serum testosterone concentrations return to baseline and gynecomastia and impotence usually abate after ketoconazole therapy is stopped. Oligospermia has been reported in patients receiving systemic ketoconazole in investigational studies. This was mainly at doses above those approved. Oligospermia was not reported at doses up to 400 mg/day; however, sperm counts are not frequently obtained at these doses.[10]
Rare cases of anaphylactoid reactions have been reported following the first dose of oral ketoconazole and during post-marketing use of the 2% shampoo. Several cases of hypersensitivity reactions including urticaria and angioedema have also been reported.[11] Fever and chills have been reported in < 1% of patients receiving systemic ketoconazole.[10] Facial swelling has been reported in < 1% of patients using the topical gel formulation.[49]
Dizziness, drowsiness (somnolence), and headache have been reported in < 1% of patients receiving treatment with oral ketoconazole. Neuropsychiatric events have also been reported rarely and include suicidal ideation and severe depression.[10] Headache and dizziness were also reported in < 1% of patients using the topical gel formulation.[49]
Photophobia occurs in < 1% of patients receiving treatment with oral ketoconazole. To minimize discomfort, the patient should wear sunglasses while outside and avoid bright light when possible.[10] Treatment-related ophthalmic adverse events reported in < 1% of patients using the topical gel include ocular irritation, ocular swelling, and keratoconjunctivitis sicca.[49]
Post-marketing worldwide reports in patients receiving oral ketoconazole have included rare cases of paresthesias, and signs of increased intracranial pressure including bulging fontanelles and papilledema. Hypertriglyceridemia has also been reported with the oral formulation but a causal relationship with ketoconazole has not been determined.[10] Paresthesias have also been reported in <= 1% of patients using the topical foam and in < 1% of patients using the topical gel.[38][49]
Dermatologic reactions were reported with systemic and topical ketoconazole formulations. Pruritus has been reported in 1.5% of patients receiving oral ketoconazole; it has also been reported following application of the topical cream, topical foam (up to 1%), topical gel (less than 1%), and the shampoo. Alopecia has been reported with the shampoo and with worldwide postmarketing reports in patients receiving the oral formulation. Skin irritation has been reported with topical use, specifically with the cream (as pruritus, burning, and stinging), the foam (up to 1%), the gel (less than 1%), and the shampoo. Contact dermatitis was reported with use of the shampoo and postmarketing with the cream; application site dermatitis was reported with use of the gel (less than 1%). The topical foam (Extina) was associated with an increased incidence of contact sensitization, including photosensitivity in dermal safety studies. Application site reaction (6%) and burning (10%) were reported with ketoconazole foam, burning was also reported with the gel (4%) and the shampoo. Skin dryness or xerosis was reported with use of the foam (up to 1%), the gel (less than 1%), and the shampoo. Erythema was reported with use of the foam (up to 1%) and the gel (less than 1%). Rash (unspecified) was reported with use of the foam (up to 1%) and the shampoo. Warmth at the application site was noted in up to 1% of patients using the foam. Pustules were noted on the skin with application of the gel (less than 1%) and on the scalp with application of the shampoo. Other reactions noted in less than 1% of patients who used the topical gel include discharge, pain, impetigo, pyogenic granuloma, acne vulgaris, and nail discoloration. Adverse reactions specifically noted with use of the shampoo include hair discoloration, abnormal hair texture and removal of curl, rash (unspecified), and application site reactions.[10][11][37][38][39]
Systemic ketoconazole can prolong the QT interval. Serious cardiovascular events, QT prolongation, ventricular arrhythmias, and torsade de pointes, have been observed in patients receiving ketoconazole oral tablets in combination with other QT-prolonging drugs.[10]
Ketoconazole alters vitamin D metabolism and may lead to vitamin D deficiency. Patients should be monitored and supplemented with vitamin D if necessary.
Minoxidil
Minoxidil is classified as pregnancy risk category C. Although no adequate human studies have examined the effects of this drug on the fetus, animal reproduction studies have shown adverse effects, including reduced ability to conceive and a reduced survival of offspring. Dysmorphic facial features and hypertrichosis were observed in an infant whose mother received a daily minoxidil dosage of 10 mg during pregnancy.[40] Therefore, in making the decision to administer this drug during pregnancy, the potential risks to the fetus and possible difficulty in conceiving must be weighed against the potential benefits to the mother.[41]
Azelaic Acid
Azelaic acid is classified FDA pregnancy risk category B. Animal data suggests embryotoxic effects when administered orally; no teratogenic effects were observed. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, azelaic acid should be used during pregnancy only if clearly needed.[4][42]
Finasteride
Finasteride is not FDA-approved for use in females of childbearing potential and is contraindicated during pregnancy. Finasteride may cause fetal harm. Finasteride and other 5-alpha-reductase inhibitors, by inhibiting the conversion of testosterone to DHT, have the ability to cause abnormalities in the external genitalia of the male fetus. Pregnant women or females trying to conceive should not handle crushed or broken finasteride tablets. The distribution of finasteride into human semen has been assessed and appears to be well below the threshold concentration associated with fetal anomalies in animals.[7][29]
Ketoconazole
There are no adequate and well-controlled studies of ketoconazole use during human pregnancy to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. Use ketoconazole in pregnant women only if the potential benefit justifies the potential risk to the fetus.[10][11][39] Guidelines recommend against starting oral azole antifungals, including ketoconazole, during pregnancy and to discontinue these agents in HIV-positive women who become pregnant.[43] Embryotoxic and teratogenic effects (syndactylia and oligodactylia) have been demonstrated in animals receiving oral ketoconazole doses at 10-times the maximum recommended human dose. In addition, dystocia was observed in animals administered oral ketoconazole during the third trimester of gestation at doses approximately one-fourth the maximum human dose, based on body surface area comparisons. Ketoconazole is not detected in human plasma after chronic shampooing of the scalp.[10][11]
Minoxidil
According to the manufacturer, minoxidil should not be administered to a nursing mother.[6800] The American Academy of Pediatrics (AAP) considers minoxidil to be generally compatible with breast-feeding; however, other experts are less comfortable with the use of this potent antihypertensive agent in nursing mothers.[44] In one case report of a woman taking minoxidil 5 mg PO twice daily, minoxidil was rapidly excreted into the breast milk. After two months, no adverse events were reported in the nursing infant. The effect of prolonged exposure during breastfeeding is unknown.[45] Examples of other antihypertensives with more data in this population that have been classified as usually compatible with breast-feeding by the AAP and may be possible alternatives for some patients include enalapril, hydrochlorothiazide, methyldopa, and propranolol.[44] It is not known whether topical minoxidil is distributed into breast milk. Consider the benefits of breastfeeding, the risk of potential infant drug exposure, and the risk of an untreated or inadequately treated condition. If a breastfeeding infant experiences an adverse effect related to a maternally administered drug, healthcare providers are encouraged to report the adverse effect to the FDA.
Azelaic Acid
According to the manufacturer, caution should be exercised when azelaic acid is administered to breastfeeding women. In vitro studies assessing human milk partitioning suggests that azelaic acid may be distributed into breast milk. However, since less than 4% of a topically applied dose is systemically absorbed, the uptake of azelaic acid into maternal milk is not expected to cause a significant change from baseline azelaic acid concentrations in the milk.[4][46] Consider the benefits of breastfeeding, the risk of potential infant drug exposure, and the risk of an untreated or inadequately treated condition. If a breastfeeding infant experiences an adverse effect related to a maternally ingested drug, healthcare providers are encouraged to report the adverse effect to the FDA.
Finasteride
Finasteride is not FDA-approved for use in females of childbearing potential and is recommended to be avoided during breastfeeding. It is not known whether finasteride is excreted in human milk. Therefore, the effects of finasteride on breastfeeding or a nursing infant cannot be determined.[7][29]
Ketoconazole
Systemic ketoconazole is excreted in breast milk. In a case report of a mother prescribed 200 mg PO daily for 10 days, ketoconazole milk concentrations of 0.22 mcg/mL (peak) were observed 3.25 hours post-dose and were undetectable at 24 hours post-dose. Assuming a milk intake of 150 mL/kg/day, the daily ketoconazole dose of an exclusively breastfed infant was calculated as 0.01 mg/kg/day or 0.4% of the mother’s weight-adjusted dose. There are no data on the effects of ketoconazole on the breastfed infant or its effects on milk production. The manufacturer recommends mothers refrain from breastfeeding their infants during oral therapy; however, previous American Academy of Pediatrics (AAP) recommendations considered ketoconazole compatible with breastfeeding. After topical application, ketoconazole concentrations in plasma are low; therefore, concentrations in human breast milk are likely to be low. Advise breastfeeding women not to apply topical ketoconazole directly to the nipple and areola to avoid direct infant exposure. Consider the developmental and health benefits of breastfeeding along with the mother’s clinical need for topical ketoconazole and any potential adverse effects on the breastfed infant from ketoconazole or the underlying maternal condition. Fluconazole may be a potential alternative to consider during breastfeeding.[10][11][38][39][44][47]
Store this medication in its original container at 68°F to 77°F (20°C to 25°C) and away from heat, moisture and light. Keep all medicine out of the reach of children. Throw away any unused medicine after the beyond use date. Do not flush unused medications or pour down a sink or drain.
- DeVillez RL. The Therapeutic Use of Topical Minoxidil. Dermatol Clin 1990;8:367-74.
- Olsen EA, Dunlap FE, Funicella T, et al. A randomized clinical trial of 5% topical minoxidil versus 2% topical minoxidil and placebo in the treatment of androgenetic alopecia in men. J Am Acad Dermatol 2002;47:377-85.
- Olsen EA, Whiting D, Bergfeld W, et al. A multicenter, randomized, placebo-controlled, double-blind clinical trial of a novel formulation of 5% minoxidil topical foam versus placebo in the treatment of androgenetic alopecia in men. J Am Acad Dermatol 2007;57(5):767-74. Epub 2007 Aug 29
- Finacea (azelaic acid) topical gel package insert. Whippany, NJ: Bayer Healthcare; 2016 Aug.
- Azelex (azelaic acid cream) 20% [package insert]. Irvine, CA: Allergan; 2013.
- Finacea (azelaic acid) 15% topical foam package insert. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc.; 2015 Jul.
- Proscar (finasteride) package insert. Whitehouse Station, NJ: Merck and Co.; 2014 Jan.
- Thompson IM, Goodman PJ, Tangen CM, et al. The influence of finasteride on the development of prostate cancer. N Engl J Med 2003:349:213-22.
- FDA Drug Safety Communication: 5-alpha reductase inhibitors (5-ARIs) may increase the risk of a more serious form of prostate cancer. Retrieved June 9, 2011. Available on the World Wide Web at: http://www.fda.gov/Drugs/DrugSafety/ucm258314.htm
- Ketoconazole tablets package insert. Morgantown, WV: Mylan Pharmaceuticals, Inc.; 2017 Sept.
- Nizoral (ketoconazole shampoo) package insert. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2017 Dec.
- Nizoral (ketoconazole) package insert. Titusville, NJ: Janssen Pharmaceutica Products, L.P.; 2006 Aug.
- Food and Drug Administration MedWatch. FDA Drug Safety Communication: FDA limits usage of Nizoral (ketoconazole) oral tablets due to potentially fatal liver injury and risk of drug interactions and adrenal gland problems. Retrieved July 26th, 2013. Available on the World Wide Web https://www.fda.gov/Drugs/DrugSafety/ucm362415.htm
- Food and Drug Administration MedWatch. FDA Drug Safety Communication: FDA warns that prescribing of Nizoral (ketoconazole) oral tablets for unapproved uses including skin and nail infections continues; linked to patient death. Retrieved May 19th, 2016. Available on the World Wide Web https://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm502073.ht
- Bauer JH, Alpert MA. Rapid reduction of severe hypertension with minoxidil. J Cardiovasc Pharmacol 1980;2 Suppl:S189-99.
- Alpert MA, Bauer JH. Rapid control of severe hypertension with minoxidil. Arch Intern Med 1982;142(12):2099-104.
- Pogatsa-Murray G, Varga L, Varga A, et al. Changes in left ventricular mass during treatment with minoxidil and cilazapril in hypertension patients with left ventricular hypertrophy. J Hum Hypertens 1997;11(3):149-56.
- Buhl AE. Minoxidil’s action in hair follicles. J Invest Dermatol 1991;96:73S-4S.
- Messenger AG, Rundegren J. Minoxidil: mechanisms of action on hair growth. Br J Dermatol 2004;150:186-194.
- Sato T, Tadokoro T, Sonoda T, et al. Minoxidil increases 17b-hydroxysteroid dehydrogenase and 5a-reductase activity of cultured human dermal papilla cells from balding scalp. J Derm Sci 1999;19:123-5.
- Como JA, Dismukes WE. Oral azole drugs as systemic antifungal therapy. N Engl J Med 1994;330:263-72.
- Borgers M. Mechanism of action of antifungal drugs, with special reference to the imidazole derivatives. Rev Inf Dis. 1980;2:520-34.
- Sonino N. The use of ketoconazole as an inhibitor of steroid production. N Engl J Med 1987;317:812-8.
- Pont A, Williams PL, Loose DS, et al. Ketoconazole blocks adrenal steroid synthesis. Ann Intern Med 1982;97:370-2.
- Trachtenberg J, Pont A. Ketoconazole therapy for advanced prostate cancer. Lancet 1984;433-5.
- Yu M, Tomasa G. A double-blind, prospective, randomized trial of ketoconazole, a thromboxane synthetase inhibitor, in the prophylaxis of the adult respiratory distress syndrome. Crit Care Med 1993;21:1635-42.
- Loniten (minoxidil) tablets package insert. Kalamazoo, MI: Pharmacia; 2006 Feb.
- Health Care Financing Administration. Interpretive Guidelines for Long-term Care Facilities. Title 42 CFR 483.25(l) F329: Unnecessary Drugs. Revised 2015.
- Propecia (finasteride) package insert. Whitehouse Station, NJ: Merck and Co., INC.; 2013 Sept.
- Amory JK, Wang C, Swerdloff RS, et al. The effect of 5-alpha-reductaxe inhibition with dutasteride and finasteride on semen parameters and serum hormones in healthy men. J Clin Endocrin Metab 2007;92(5):1659-65
- Roden, DM. Drug-induced prolongation of the QT interval. New Engl J Med 2004;350:1013-22.
- Crouch MA, Limon L, Cassano AT. Clinical relevance and management of drug-related QT interval prolongation. Pharmacotherapy 2003;23:881-908.
- Benoit SR, Mendelsohn AB, Nourjah P, et al. Risk factors for prolonged QTc among US adults: Third National Health and Nutrition Examination Survey. Eur J Cardiovasc Prev Rehabil 2005;12(4):363-368.
- Koide T, Ozeki K, Kaihara S, et al. Etiology of QT prolongation and T wave changes in chronic alcoholism. Jpn Heart J 1981;22:151-166.
- van Noord C, Eijgelsheim M, Stricker BH. Drug- and non-drug-associated QT interval prolongation. Br J Clin Pharmacol 2010;70(1):16-23.
- Galli-Tsinopoulou A, Chatzidimitriou A, Kyrgios I, et al. Children and adolescents with type 1 diabetes mellitus have a sixfold greater risk for prolonged QTc interval. J Pediatr Endocrinol Metab 2014;27:237-243.
- Ketoconazole cream package insert. Suwanee, GA: Tiber Laboratories; 2010 Apr.
- Extina (ketoconazole foam) package insert. Morgontown, WV: Mylan Pharmaceuticals, Inc.; 2018 Jun.
- Xolegel (ketoconazole gel) package insert. Exton, PA: Almirall, LLC; 2019 Dec.
- Kaler SG, Patrinos ME, Lambert GH, et al. Hypertrichosis and congenital anomalies associated with maternal use of minoxidil. Pediatrics 1987;79:434-6.
- Minoxidil tablets package insert. Corona, CA: Watson Laboratories, Inc; 2009 Jun.
- Azelex (azelaic acid) package insert. Irvine, CA: Allergan, Inc.; 2004 May.
- US Public Health Service (USPHS) and the Infectious Diseases Society of America (IDSA). The Living Document: Guidelines for the Preventing Opportunistic Infections Among HIV-Infected Persons. Retrieved November 28, 2001. Available on the World Wide Web at www.aidsinfo.nih.gov.
- American Academy of Pediatrics (AAP) Committee on Drugs. Transfer of drugs and other chemicals into human milk. Pediatrics 2001;108(3):776-789.
- Valdivieso A, Valdes G, Spiro TE, et al. Minoxidil in breast milk. Ann Intern Med. 1985;102:135. Letter.
- Azelex (azelaic acid) package insert. Irvine, CA: Allergan, Inc.; 2004 May.
- Moretti ME, Ito S, Koren G. Disposition of maternal ketoconazole in breast milk. Am J Obstet Gynecol. 1995;173:1625-1626.
- Green L, Wysowski DK, Fourcroy JL. Gynecomastia and breast cancer during finasteride therapy. N Engl J Med 1996;335:823.
- Xolegel (ketoconazole gel) package insert. Exton, PA: Almirall, LLC; 2019 Dec.
Administration Instructions
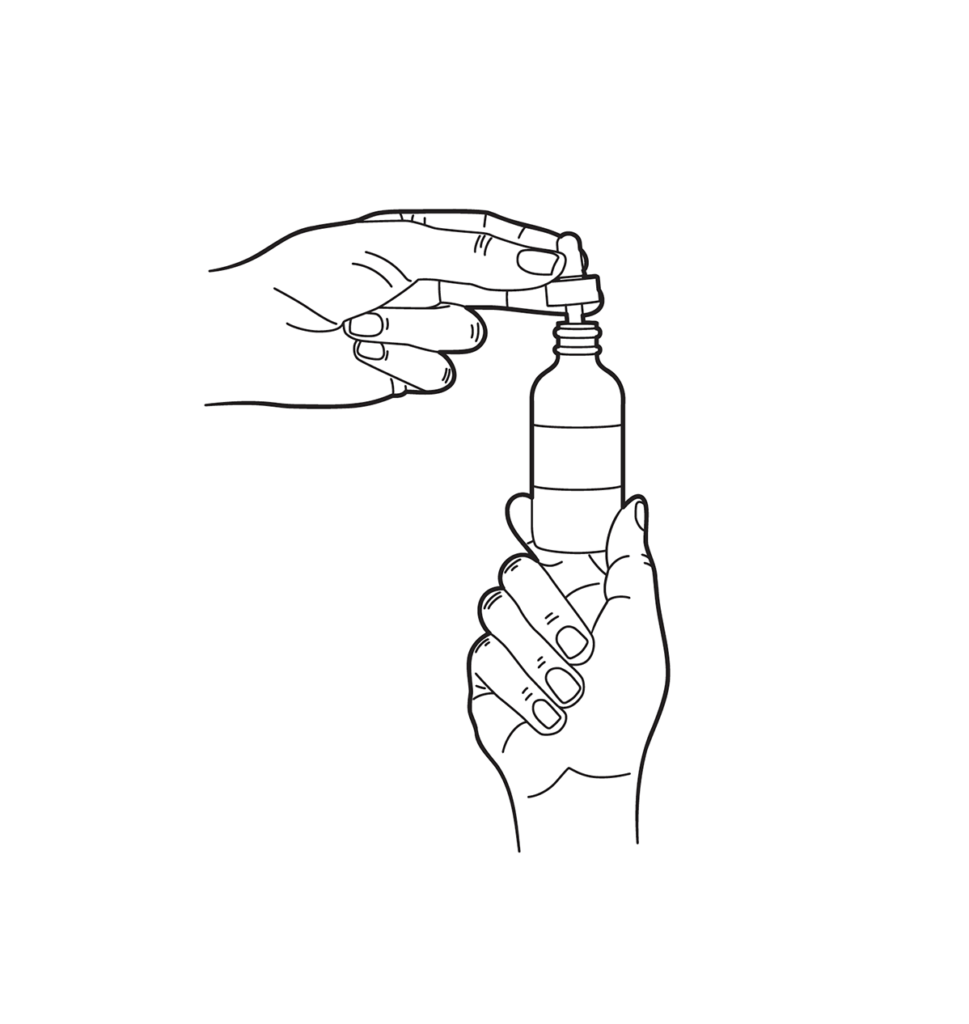
Dropper Instructions
 GHK-Cu Scalp Solution
GHK-Cu Scalp Solution Hair Restore D Scalp Solution
Hair Restore D Scalp Solution Hair Restore Extreme Scalp Solution
Hair Restore Extreme Scalp Solution Minoxidil Capsules
Minoxidil Capsules Dutasteride Capsules
Dutasteride Capsules Finasteride Tablets
Finasteride Tablets Biotin / Minoxidil / Spironolactone Capsules
Biotin / Minoxidil / Spironolactone Capsules Biotin / Minoxidil Capsules
Biotin / Minoxidil Capsules Biotin (Vitamin B7) Injection
Biotin (Vitamin B7) Injection Tirzepatide / Niacinamide Injection
Tirzepatide / Niacinamide Injection503A vs 503B
- 503A pharmacies compound products for specific patients whose prescriptions are sent by their healthcare provider.
- 503B outsourcing facilities compound products on a larger scale (bulk amounts) for healthcare providers to have on hand and administer to patients in their offices.
Frequently asked questions
Our team of experts has the answers you're looking for.
A clinical pharmacist cannot recommend a specific doctor. Because we are licensed in all 50 states*, we can accept prescriptions from many licensed prescribers if the prescription is written within their scope of practice and with a valid patient-practitioner relationship.
*Licensing is subject to change.
Each injectable IV product will have the osmolarity listed on the label located on the vial.

Given the vastness and uniqueness of individualized compounded formulations, it is impossible to list every potential compound we offer. To inquire if we currently carry or can compound your prescription, please fill out the form located on our Contact page or call us at (877) 562-8577.
We source all our medications and active pharmaceutical ingredients from FDA-registered suppliers and manufacturers.