

Hydrocortisone Tablets
Product Overview
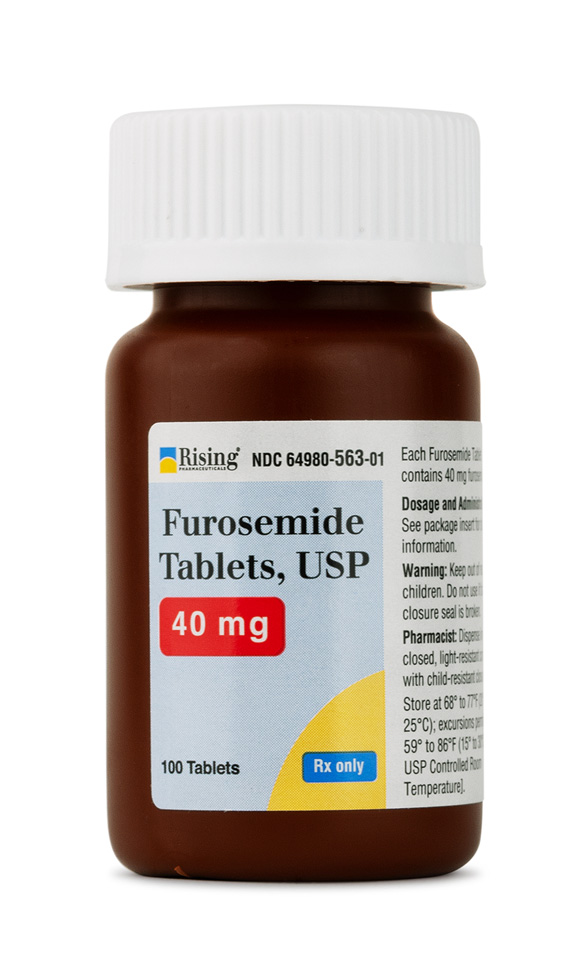 Furosemide Tablets
Furosemide Tablets Hydrochlorothiazide / Triamterene Tablets
Hydrochlorothiazide / Triamterene Tablets Hydrochlorothiazide Tablets
Hydrochlorothiazide Tablets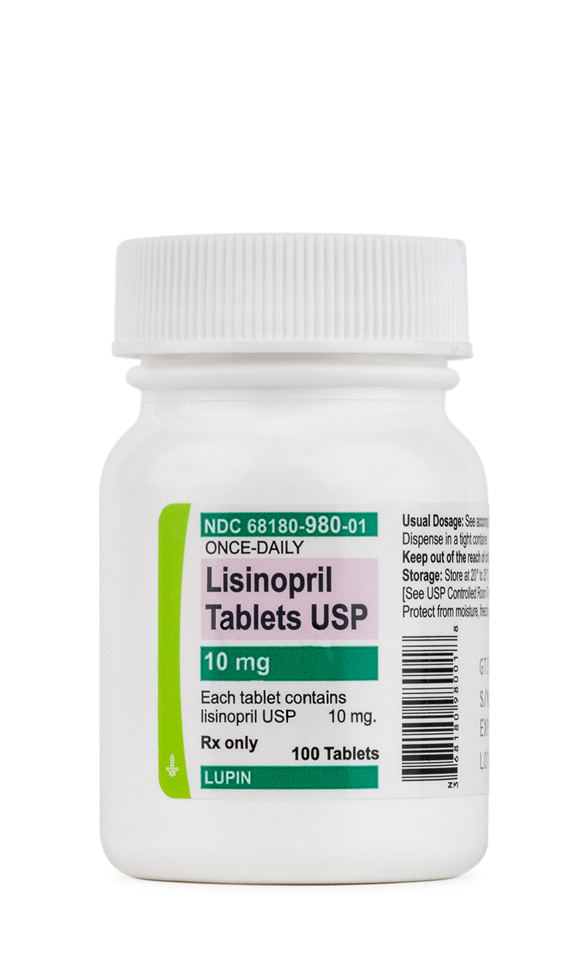 Lisinopril Tablets
Lisinopril Tablets Tirzepatide / Niacinamide Injection
Tirzepatide / Niacinamide Injection Cyanocobalamin (Vitamin B12) Injection
Cyanocobalamin (Vitamin B12) Injection503A vs 503B
- 503A pharmacies compound products for specific patients whose prescriptions are sent by their healthcare provider.
- 503B outsourcing facilities compound products on a larger scale (bulk amounts) for healthcare providers to have on hand and administer to patients in their offices.
Frequently asked questions
Our team of experts has the answers you're looking for.
